Home » Медтехника » Требования и процесс сертификации для экспорта медицинского оборудования в Австралию
Австралия располагает развитой и обширной системой общественного и частного здравоохранения, котораяМедтехникатребования также весьма высоки. Однако, чтобы продавать, импортировать или экспортировать медицинское оборудование в Австралии, необходимо пройти регистрацию и аудит в соответствии с соответствующими правилами и получить сертификат допуска. В этой статье этот процесс будет подробно описан.
Уже в 1966 году Австралия приняла "Закон о медицинских изделиях" для регулирования медицинских изделий. Сейчас основными регулирующими актами являются "Закон о медицинских изделиях" 1989 года, а также "Правила медицинских приборов", принятые в 2002 году. Исполнение и контроль за этими двумя нормативными актами осуществляет подведомственный орган Министерства здравоохранения и социального обеспечения пожилых людей Австралии — TGA (Therapeutic Goods Administration). Поэтому, чтобы продавать на территорию Австралии,Импорт и экспортПроизводители медицинского оборудования должны подать заявление на получение разрешения на вход на рынок в ТГА (Therapeutic Goods Administration).
Второе: Определение и классификация медицинского оборудования Австралии
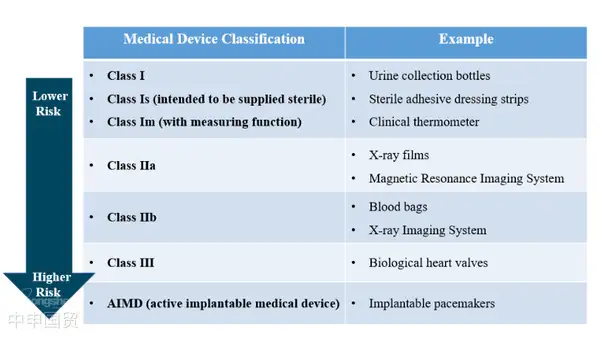
Австралия определяет медицинское оборудование как лечебный препарат, состоящий из инструментов, приборов, принадлежностей или других изделий, а также необходимых приспособлений или программного обеспечения. Эти лечебные препараты достигают основного действия преимущественно нелекарственным, иммунологическим или метаболическим путем, хотя эти способы могут способствовать их действию. В зависимости от уровня риска медицинское оборудование подразделяется на категории I, IIa, IIb, III, а также AIMD (активное имплантируемое медицинское оборудование).
III. Маркетинговое допускание медицинского оборудования в Австралии
TGA классифицирует медицинские изделия на три категории для регулирования: освобождение от регистрации, регистрация и запись. Каждой категории медицинских изделий необходимо получить правительственное одобрение перед продажей на рынке и соответствовать базовым требованиям к медицинским изделиям. Высокорисковые медицинские изделия должны пройти оценку TGA и получить одобрение перед введением на рынок. В то время как низкорисковые медицинские изделия могут оцениваться предприятиями самостоятельно и могут входить на рынок, если они соответствуют требованиям к качеству и безопасности. Все медицинские изделия, производимые в Австралии, должны соответствовать стандартам GMP и производиться в чистой и неконтаминированной среде.
IV. Анализ процесса регистрации медицинского оборудования в Австралии
Процесс введения медицинского оборудования на australийский рынок — это тщательная и трудоемкая работа, которая требует выполнения обязанностей нескольких ключевых участников, четкого определения медицинского оборудования и его уровня риска, а также прохождения оценки соответствия и регистрации в ARTG.
(1)Уточнить ключевые роли
Сначала процесс регистрации и ввода медицинского оборудования на australийский рынок затрагивает две ключевые роли: производителя (Manufacturer) и спонсора (Sponsor). Производитель отвечает за дизайн, производство, упаковку, отправку медицинского оборудования и т.д., а спонсор — это гражданин Австралии или австралийская компания, которая занимается получением регистрационного номера спонсора в TGA (Терапевтическое Управление Товарами) для помощи в завершении процесса регистрации.
(2)明確醫(yī)療器械的定義及風(fēng)險(xiǎn)等級(jí): уточнить определение медицинского оборудования и его категорию риска
Далее необходимо明確 определить определение и уровень риска медицинских устройств. Определение и основные моменты медицинских устройств в "Законе о медицинских изделиях" 1989 года следующие:

В Австралии медицинские устройства, соответствующие определению, данному в ?Законе о медицинских изделиях 1989 года?, должны быть зарегистрированы в TGA (Therapeutic Goods Administration). В зависимости от степени риска и предполагаемого применения медицинские устройства разделены на пять категорий риска: чем выше категория риска, тем более строгая регулировка со стороны TGA.
(3)Заявление на получение свидетельства о соответствии
Перед регистрацией и вводом медицинского оборудования на рынок производитель должен провести соответствующую процедуру оценки соответствия, пройти проверку ТГА и получить действующее свидетельство о проведении оценки соответствия. Свидетельства о проведении оценки соответствия, признанные ТГА, включают TGA Conformity Assessment Evidence, EC Certificate, а также сертификаты, выданные на основе EC-MRA и EFTA-MRA.
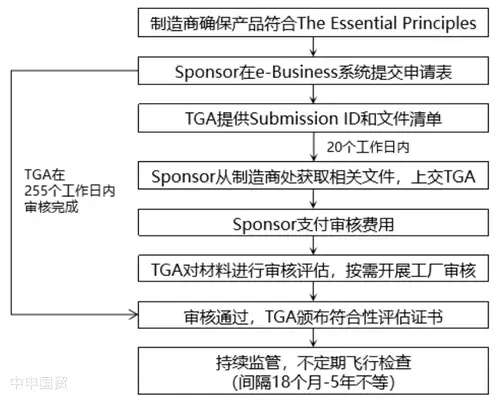
Если для оценки соответствия выбирается доказательство соответствия TGA, процесс подачи заявки на сертификат следующий:
(4)Предоставить регистрацию ARTG
После завершения оценки соответствия изготовитель и спонсор должны подать заявление на регистрацию в ARTG для получения сертификата допуска для отдельного медицинского изделия с целью завершения внесения продукта в реестр. Только после внесения медицинского изделия в Регистр терапевтических товаров Австралии (ARTG) оно может быть реализовано на australianском рынке.

ПЯТЬ. Система качества медицинского оборудования Австралии
Правительство установило, что производственный процесс всех предприятий, занимающихся производством медицинского оборудования, должен соответствовать требованиям к качеству, связанным с производимым оборудованием, а также обладать средствами и процедурами обеспечения качества. Для достижения этой цели Австралия внедрила систему добросовестного производства (GMP) и в то же время ориентируется на стандарты системы качества ISO 9000. С 2017 года Австралия начала применять единый процедуру аудита медицинского оборудования и принимать сертификаты MDSAP, выданные квалифицированными проверочными организациями.
Шесть. Постмаркетинговое управление медицинскими изделиями в Австралии
Это достигается главным образом через систему послемаркетингового контроля. Австралия, посредством отчетов о неблагоприятных явлениях, лабораторных испытаний и мониторинга товаров, поступивших на рынок, гарантирует, что все медицинские устройства после ввода на рынок соответствуют требованиям регулирующих документов. Ее правила мониторинга неблагоприятных явлений после выхода на рынок очень зрелые, процедуры подробные, сочетающие принципиальность и гибкость, и обладают сильной оперативностью.
VII. Правила изменения регистрации медицинских изделий в Австралии
После того как продукт получит разрешение на поступление на рынок, его информация и статус должны продолжать регулироваться. Если продукт подвергся существенным изменениям, производитель и спонсор должны своевременно уведомить Управление по регулированию терапевтической продукции (TGA). Спонсор должен войти в систему e-Business и подать заявление на изменение регистрации. Такие изменения могут включать изменения в информации о производителе, спонсоре, продукте, изменения в информации о сертификате соответствия, изменения предполагаемого назначения продукта, клинических показаний, а также изменения категории, к которой относится продукт, и кода GMDN.
Итак, чтобы продавать, импортировать или экспортировать медицинское оборудование в Австралии, необходимо пройти ряд строгих процедур допуска и регистрации. Эти процедуры гарантируют качество, безопасность и эффективность медицинского оборудования, а следовательно, и качество системы здравоохранения.
Рекомендуем также:






? 2025. All Rights Reserved. 滬ICP備2023007705號(hào)-2  Номер разрешения на безопасность в сети Шанхая: 31011502009912.
Номер разрешения на безопасность в сети Шанхая: 31011502009912.